import Bio
print(Bio.__version__)1.86Python offers a variety of functions to work with text data (Strings) that in turn make it easier to work with biological data such as DNA or protein sequences. BioPython library provides a set of classes dedicated to parsing and analysis of different type of biological data. The functions avaiable in BioPython helps researcher to progammatically process the data. Below we’ll see some of the features in Biopython for working with biological data.
To install Biopython library run pip install biopython. For more details regarding Biopython installation and tutorials, please refer to the Biopython wiki.
To check the version of Biopython, run the following command.
import Bio
print(Bio.__version__)1.86To work with sequences, we’ll need the Bio.Seq class which has the required functions for reading and writing sequence data. Once we have imported this class we can create objects having required data. The example below shows constructing a sequence object with a DNA sequence and then using the complement and translate functions to find the sequence of the complementary strand and the translated protein sequence, respectively.
from Bio.Seq import Seq
new_sequence = Seq('AATTGGAACCTT')
print(new_sequence)
print(new_sequence.complement())
print(new_sequence.translate())Looking at the sequence object, one might wonder how is this different from a string object. Because we can easily initialize a string variable with the required sequence as the value. The difference here is that the two are the objects of different classes i.e. the Seq object belongs to the Bio.Seq.Seq class whereas the string object belongs to Str. These differences in turn indicate the differences in the kind of functionality associated with these objects (think object-oriented programming). For example, the complement and translate functions make sense only in the case of sequences and not in the case of any generic text.
The Bio.SeqUtils package offers a set of utility functions for getting some basic information about the sequences. These include, e.g., calculating the GC content or the melting temperature for the DNA sequences.
from Bio.SeqUtils import gc_fraction
from Bio.SeqUtils import MeltingTemp as mt
print(gc_fraction(new_sequence))
print(mt.Tm_Wallace(new_sequence)) # Tm_GC can also be used.Similarly, there are functions to get information about the protein sequences. The ProtParam module has utility functions for performing basic analysis of protein sequences. For example, the molecular_weight function returns the molecular weight of the protein and the count_amino_acids function returns a dictionary having the count for all the amino acids.
from Bio.SeqUtils.ProtParam import ProteinAnalysis
P1 = ProteinAnalysis("MFAEGRNREST")
print(P1.molecular_weight())
print(P1.count_amino_acids()) # get_amino_acid_percent can also be used.When we are working with the sequence data, we generally do not type sequences instead we read (and write) sequences from a file. We’ll now see how to read and write sequences using BioPython.
To create a sequence object by reading sequence from a file, we can use the SeqIO class. The parse function in this class can read and write sequences in different formats. This function take two arguments - file name and format, and return an iterator having all the sequences. The code below shows reading a file having multiple sequences and printing the sequences using the seq attribute. Note that this would print sequnces without any annotations. The description attribute of SeqIO object can used to print the description of a sequence as given in the input file. In the code below, the output would be restricted to first three sequences. If you need to print the data for all sequences then remove the if block. The sequence file used in the example below is available here
from Bio import SeqIO
count=0
for all_seqs in SeqIO.parse("Human_MAPK.fasta", "fasta"):
print(all_seqs.description)
print(all_seqs.seq)
count += 1
if(count == 3):
breaksp|O15264|MK13_HUMAN Mitogen-activated protein kinase 13 OS=Homo sapiens OX=9606 GN=MAPK13 PE=1 SV=1
MSLIRKKGFYKQDVNKTAWELPKTYVSPTHVGSGAYGSVCSAIDKRSGEKVAIKKLSRPFQSEIFAKRAYRELLLLKHMQHENVIGLLDVFTPASSLRNFYDFYLVMPFMQTDLQKIMGMEFSEEKIQYLVYQMLKGLKYIHSAGVVHRDLKPGNLAVNEDCELKILDFGLARHADAEMTGYVVTRWYRAPEVILSWMHYNQTVDIWSVGCIMAEMLTGKTLFKGKDYLDQLTQILKVTGVPGTEFVQKLNDKAAKSYIQSLPQTPRKDFTQLFPRASPQAADLLEKMLELDVDKRLTAAQALTHPFFEPFRDPEEETEAQQPFDDSLEHEKLTVDEWKQHIYKEIVNFSPIARKDSRRRSGMKL
sp|O60271|JIP4_HUMAN C-Jun-amino-terminal kinase-interacting protein 4 OS=Homo sapiens OX=9606 GN=SPAG9 PE=1 SV=4
MELEDGVVYQEEPGGSGAVMSERVSGLAGSIYREFERLIGRYDEEVVKELMPLVVAVLENLDSVFAQDQEHQVELELLRDDNEQLITQYEREKALRKHAEEKFIEFEDSQEQEKKDLQTRVESLESQTRQLELKAKNYADQISRLEEREAELKKEYNALHQRHTEMIHNYMEHLERTKLHQLSGSDQLESTAHSRIRKERPISLGIFPLPAGDGLLTPDAQKGGETPGSEQWKFQELSQPRSHTSLKVSNSPEPQKAVEQEDELSDVSQGGSKATTPASTANSDVATIPTDTPLKEENEGFVKVTDAPNKSEISKHIEVQVAQETRNVSTGSAENEEKSEVQAIIESTPELDMDKDLSGYKGSSTPTKGIENKAFDRNTESLFEELSSAGSGLIGDVDEGADLLGMGREVENLILENTQLLETKNALNIVKNDLIAKVDELTCEKDVLQGELEAVKQAKLKLEEKNRELEEELRKARAEAEDARQKAKDDDDSDIPTAQRKRFTRVEMARVLMERNQYKERLMELQEAVRWTEMIRASRENPAMQEKKRSSIWQFFSRLFSSSSNTTKKPEPPVNLKYNAPTSHVTPSVKKRSSTLSQLPGDKSKAFDFLSEETEASLASRREQKREQYRQVKAHVQKEDGRVQAFGWSLPQKYKQVTNGQGENKMKNLPVPVYLRPLDEKDTSMKLWCAVGVNLSGGKTRDGGSVVGASVFYKDVAGLDTEGSKQRSASQSSLDKLDQELKEQQKELKNQEELSSLVWICTSTHSATKVLIIDAVQPGNILDSFTVCNSHVLCIASVPGARETDYPAGEDLSESGQVDKASLCGSMTSNSSAETDSLLGGITVVGCSAEGVTGAATSPSTNGASPVMDKPPEMEAENSEVDENVPTAEEATEATEGNAGSAEDTVDISQTGVYTEHVFTDPLGVQIPEDLSPVYQSSNDSDAYKDQISVLPNEQDLVREEAQKMSSLLPTMWLGAQNGCLYVHSSVAQWRKCLHSIKLKDSILSIVHVKGIVLVALADGTLAIFHRGVDGQWDLSNYHLLDLGRPHHSIRCMTVVHDKVWCGYRNKIYVVQPKAMKIEKSFDAHPRKESQVRQLAWVGDGVWVSIRLDSTLRLYHAHTYQHLQDVDIEPYVSKMLGTGKLGFSFVRITALMVSCNRLWVGTGNGVIISIPLTETNKTSGVPGNRPGSVIRVYGDENSDKVTPGTFIPYCSMAHAQLCFHGHRDAVKFFVAVPGQVISPQSSSSGTDLTGDKAGPSAQEPGSQTPLKSMLVISGGEGYIDFRMGDEGGESELLGEDLPLEPSVTKAERSHLIVWQVMYGNE
sp|O60336|MABP1_HUMAN Mitogen-activated protein kinase-binding protein 1 OS=Homo sapiens OX=9606 GN=MAPKBP1 PE=1 SV=4
MAVEGSTITSRIKNLLRSPSIKLRRSKAGNRREDLSSKVTLEKVLGITVSGGRGLACDPRSGLVAYPAGCVVVLFNPRKHKQHHILNSSRKTITALAFSPDGKYLVTGESGHMPAVRVWDVAEHSQVAELQEHKYGVACVAFSPSAKYIVSVGYQHDMIVNVWAWKKNIVVASNKVSSRVTAVSFSEDCSYFVTAGNRHIKFWYLDDSKTSKVNATVPLLGRSGLLGELRNNLFTDVACGRGKKADSTFCITSSGLLCEFSDRRLLDKWVELRNIDSFTTTVAHCISVSQDYIFCGCADGTVRLFNPSNLHFLSTLPRPHALGTDIASVTEASRLFSGVANARYPDTIALTFDPTNQWLSCVYNDHSIYVWDVRDPKKVGKVYSALYHSSCVWSVEVYPEVKDSNQACLPPSSFITCSSDNTIRLWNTESSGVHGSTLHRNILSSDLIKIIYVDGNTQALLDTELPGGDKADASLLDPRVGIRSVCVSPNGQHLASGDRMGTLRVHELQSLSEMLKVEAHDSEILCLEYSKPDTGLKLLASASRDRLIHVLDAGREYSLQQTLDEHSSSITAVKFAASDGQVRMISCGADKSIYFRTAQKSGDGVQFTRTHHVVRKTTLYDMDVEPSWKYTAIGCQDRNIRIFNISSGKQKKLFKGSQGEDGTLIKVQTDPSGIYIATSCSDKNLSIFDFSSGECVATMFGHSEIVTGMKFSNDCKHLISVSGDSCIFVWRLSSEMTISMRQRLAELRQRQRGGKQQGPSSPQRASGPNRHQAPSMLSPGPALSSDSDKEGEDEGTEEELPALPVLAKSTKKALASVPSPALPRSLSHWEMSRAQESVGFLDPAPAANPGPRRRGRWVQPGVELSVRSMLDLRQLETLAPSLQDPSQDSLAIIPSGPRKHGQEALETSLTSQNEKPPRPQASQPCSYPHIIRLLSQEEGVFAQDLEPAPIEDGIVYPEPSDNPTMDTSEFQVQAPARGTLGRVYPGSRSSEKHSPDSACSVDYSSSCLSSPEHPTEDSESTEPLSVDGISSDLEEPAEGDEEEEEEEGGMGPYGLQEGSPQTPDQEQFLKQHFETLASGAAPGAPVQVPERSESRSISSRFLLQVQTRPLREPSPSSSSLALMSRPAQVPQASGEQPRGNGANPPGAPPEVEPSSGNPSPQQAASVLLPRCRLNPDSSWAPKRVATASPFSGLQKAQSVHSLVPQERHEASLQAPSPGALLSREIEAQDGLGSLPPADGRPSRPHSYQNPTTSSMAKISRSISVGENLGLVAEPQAHAPIRVSPLSKLALPSRAHLVLDIPKPLPDRPTLAAFSPVTKGRAPGEAEKPGFPVGLGKAHSTTERWACLGEGTTPKPRTECQAHPGPSSPCAQQLPVSSLFQGPENLQPPPPEKTPNPMECTKPGAALSQDSEPAVSLEQCEQLVAELRGSVRQAVRLYHSVAGCKMPSAEQSRIAQLLRDTFSSVRQELEAVAGAVLSSPGSSPGAVGAEQTQALLEQYSELLLRAVERRMERKLThe write function takes three arguments — 1) a sequence object, 2) filename, and 3) file format. The code below reads a fasta file with multiple sequences and then save the first 10 sequences in a new file.
all_seqs = []
for seq_record in SeqIO.parse("Human_MAPK.fasta", "fasta"):
all_seqs.append(seq_record)
print(len(all_seqs))
SeqIO.write(all_seqs[0:5],"first_5.fasta","fasta")The AlignIO class has functions to parse alignment files. The read and write functions have a similar syntax to the corresponding functions in the SeqIO class. The alignment object stores sequences in 2D array format such that the rows are number of sequences and columns represent alignment length. To extract a sub-set of an alignment, slicing feature can be used. The code below shows reading an alignment file in fasta format followed by selecting a portion of this alignment and save it in a new file in clustal format. The subset is extracted by giving the range for the rows and columns within square brackets. The numbering for both rows and columns starts from zero. In the example below first ten sequences in the alignment are selected since range of rows is :10 and the colums range is 3:12. The sequence file used in the code below is available here.
from Bio import AlignIO
align1 = AlignIO.read("kinases.txt", "fasta")
# slicing - [row range, col range]
x = align1[:10,3:12]
print(x)
AlignIO.write(x,"msa1.aln","clustal")# %load msa1.aln
CLUSTAL X (1.81) multiple sequence alignment
consensus LKVL-GKGA
sp|Q22RR1||agc:agc-sar|Tetrahy IKEL-GRGN
sp|Q234E6||agc:agc-sar|Tetrahy IKKL-GFGQ
sp|Q23KG5||agc:agc-sar|Tetrahy VKKL-GNGQ
sp|Q23DN8||agc:agc-sar|Tetrahy IKTL-AFGQ
sp|I7MFS4||agc:agc-sar|Tetrahy IKKL-GVGQ
sp|I7M3B5||agc:agc-sar|Tetrahy IKKL-GFGQ
sp|I7MD55||agc:agc-sar|Tetrahy IKKL-GFGQ
sp|Q869J9|pkg-2|agc:pkg|Parame IKKL-GEGQ
sp|A8N3F0||agc:agc-unique|Copr IRVL-GKGC
from Bio import Entrez, SeqIO
Entrez.email = "you@example.com"
handle1 = Entrez.efetch(db="nucleotide", id="NM_001301717", rettype="gb", retmode="text")
record1 = SeqIO.read(handle1, "genbank")print(record1.description)
print(len(record1.seq))
print(record1.seq[:50]) To retrieve multiple sequence, pass a comma-separated ids to the id keyword parameter. This time we’ll need to use the parse function to get the sequences from the efetch output.
all_ids = ["NP_000240.1", "NP_000549.1", "NP_001289656.1"]
handle2 = Entrez.efetch(db="protein", id=",".join(all_ids), rettype="gb", retmode="text")
record2 = list(SeqIO.parse(handle2, "genbank"))print(f"There are {len(record2)} sequences")
for record in record2:
print(record.description)
print(len(record.seq))import requests
uniprot_id = "P12345" # Replace with the protein UniProt ID
url = f"https://www.uniprot.org/uniprot/{uniprot_id}.fasta"
response = requests.get(url)
print(response.text)To get multiple sequences from Uniprot use the bulk query format
uniprot_ids = ["P69905", "P68871"]
query = " OR ".join([f"accession:{uid}" for uid in uniprot_ids])
url = f"https://rest.uniprot.org/uniprotkb/stream?format=fasta&query={query}"
response = requests.get(url)
response.raise_for_status() # Raise an error for failed requestsfrom io import StringIO
fasta_io = StringIO(response.text)
records = list(SeqIO.parse(fasta_io, "fasta"))for record in records:
print(record.description)
print(len(record.seq))Biopython offers a functionality to programmatically run BLAST on the NCBI servers using the Bio.Blast class. To run blast online at NCBI servers, Bio.Blast can be used which has different function to run Blast and also to parse the output. The qblast function takes atleast three arguments &emdash; 1) blast program (blastp, blastn, etc.), 2) database (any of the databases available at NCBI, and 3) sequence. Once the blast serach is over the output can be saved in a file. This output would be in the XML format. We can use the read or parse functions to process this output. The code below shows running a blast search using qblast against the non-redundant database available at NCBI. The output file has all the hits identified in the Blast search. These hits follow a hierarchical manner such that each result will have multiple alignments and within each alignment there will be multiple high scoring pairs (hsps) i.e. Blast object \(\longrightarrow\) hits \(\longrightarrow\) aligments. For more details on this you may refer to the Blast documentation available at NCBI.
from Bio import Blast
Blast.email = "test@example.com"
print(Blast.email)test@example.comresults_blast = Blast.qblast("blastn", "nt", "8332116")
with open("my_blast.xml", "wb") as file_handle:
file_handle.write(results_blast.read())
results_blast.close()blast_record = Blast.read("my_blast.xml")
print("Number of hits:", len(blast_record))
print(blast_record[:3])Number of hits: 50
Program: BLASTN 2.17.0+
db: core_nt
Query: BE037100.1 (length=1111)
MP14H09 MP Mesembryanthemum crystallinum cDNA 5' similar to cold
acclimation protein, mRNA sequence
Hits: ---- ----- ----------------------------------------------------------
# # HSP ID + description
---- ----- ----------------------------------------------------------
0 1 gi|1219041180|ref|XM_021875076.1| PREDICTED: Chenopodi...
1 1 gi|2514617377|ref|XM_021992092.2| PREDICTED: Spinacia ...
2 1 gi|2518612504|ref|XM_010682658.3| PREDICTED: Beta vulg...print(blast_record.keys())['gi|1219041180|ref|XM_021875076.1|', 'gi|2514617377|ref|XM_021992092.2|', 'gi|2518612504|ref|XM_010682658.3|', 'gi|2031543140|ref|XM_041168865.1|', 'gi|2618480339|ref|XM_048479995.2|', 'gi|2082357253|ref|XM_043119041.1|', 'gi|2082357255|ref|XM_043119049.1|', 'gi|1882610310|ref|XM_035691634.1|', 'gi|1882610309|ref|XM_018970776.2|', 'gi|1350315636|ref|XM_006425716.2|', 'gi|2395983799|ref|XM_006466625.3|', 'gi|2395983797|ref|XM_006466624.4|', 'gi|1204884098|ref|XM_021445554.1|', 'gi|2395983798|ref|XM_006466623.4|', 'gi|1350315638|ref|XM_006425719.2|', 'gi|2395983800|ref|XM_006466626.4|', 'gi|1350315641|ref|XM_024180293.1|', 'gi|1350315634|ref|XM_006425717.2|', 'gi|2395983796|ref|XM_025094967.2|', 'gi|1227938481|ref|XM_022049453.1|', 'gi|1063463253|ref|XM_007047033.2|', 'gi|1063463252|ref|XM_007047032.2|', 'gi|1269881403|ref|XM_022895603.1|', 'gi|1269881407|ref|XM_022895605.1|', 'gi|1269881405|ref|XM_022895604.1|', 'gi|2082386146|ref|XM_043113302.1|', 'gi|2082386143|ref|XM_043113301.1|', 'gi|1954740698|ref|XM_038867092.1|', 'gi|1882636119|ref|XM_018974650.2|', 'gi|2526866810|ref|XM_057645500.1|', 'gi|1187397285|gb|KX009413.1|', 'gi|2550782781|ref|XM_058372567.1|', 'gi|2806124758|ref|XM_068481225.1|', 'gi|2532162279|ref|XM_058104265.1|', 'gi|2250518185|ref|XM_009343631.3|', 'gi|1350280614|ref|XM_024170292.1|', 'gi|743838297|ref|XM_011027373.1|', 'gi|743838293|ref|XM_011027372.1|', 'gi|1768569081|ref|XM_031406607.1|', 'gi|2396494060|ref|XM_052454347.1|', 'gi|2396494064|ref|XM_024605027.2|', 'gi|1585724761|ref|XM_028202722.1|', 'gi|2537663858|ref|XM_021815584.2|', 'gi|2960782598|ref|XM_035077206.2|', 'gi|2645357626|ref|XM_062094449.1|', 'gi|1162571919|ref|XM_020568695.1|', 'gi|1162571918|ref|XM_007202530.2|', 'gi|2583747300|ref|XM_059787294.1|', 'gi|1229761331|ref|XM_022277554.1|', 'gi|2118882425|ref|XM_044613294.1|']print('gi|2514617377|ref|XM_021992092.2|' in blast_record)Trueprint(type(blast_record[0]))
print(blast_record[0])<class 'Bio.Blast.Hit'>
Query: BE037100.1
MP14H09 MP Mesembryanthemum crystallinum cDNA 5' similar to cold
acclimation protein, mRNA sequence
Hit: gi|1219041180|ref|XM_021875076.1| (length=1173)
PREDICTED: Chenopodium quinoa cold-regulated 413 plasma membrane protein
2-like (LOC110697660), mRNA
HSPs: ---- -------- --------- ------ --------------- ---------------------
# E-value Bit score Span Query range Hit range
---- -------- --------- ------ --------------- ---------------------
0 6.4e-117 435.90 624 [58:678] [277:901]hit1 = blast_record[0]
print(type(hit1[0]))
print(hit1[0])<class 'Bio.Blast.HSP'>
Query : BE037100.1 Length: 1111 Strand: Plus
MP14H09 MP Mesembryanthemum crystallinum cDNA 5' similar to cold
acclimation protein, mRNA sequence
Target: gi|1219041180|ref|XM_021875076.1| Length: 1173 Strand: Plus
PREDICTED: Chenopodium quinoa cold-regulated 413 plasma membrane protein
2-like (LOC110697660), mRNA
Score:435 bits(482), Expect:6e-117,
Identities:473/624(76%), Positives:473/624(76%), Gaps:4.624(1%)
gi|121904 277 ACCGAAAATGGGCAGAGGAGTGAATTATATGGCAATGACACCTGAGCAACTAGCCGCGGC
0 ||.|||||||||.||||.|.||||.||..||||.||||.|.||||.|||.|.||||.|||
BE037100. 58 ACAGAAAATGGGGAGAGAAATGAAGTACTTGGCCATGAAAACTGATCAATTGGCCGTGGC
gi|121904 337 CAATTTGATCAACTCCGACATCAATGAGCTCAAGATCGTTGTGATGACACTCATTCATGA
60 .|||.|||||.|.|||||.|||||||||||.||.||.|.....||||..|||||..||||
BE037100. 118 TAATATGATCGATTCCGATATCAATGAGCTTAAAATGGCAACAATGAGGCTCATCAATGA
gi|121904 397 TGCTTCTAGACTCGGCGGCACCTCAGGATTTGGAACTCATTTTCTTAGATGGCTAGCCTC
120 ||||..||..|||||.--||...|.||-|||||.||||||||.||.|.||||||.||||.
BE037100. 178 TGCTAGTATGCTCGGT--CATTACGGG-TTTGGCACTCATTTCCTCAAATGGCTCGCCTG
gi|121904 457 TCTTGCTGCTATTTACTTGTTGATCCTGGATCGCACAAATTGGAGAACCAACATGCTCAC
180 .|||||.|||||||||||||||||..|||||||.|||||.||||||||||||||||||||
BE037100. 235 CCTTGCGGCTATTTACTTGTTGATATTGGATCGAACAAACTGGAGAACCAACATGCTCAC
gi|121904 517 ATCACTCTTAGTACCATACATATTCCTCAGTCTTCCTTCTGGCCCTTTTTACCTTCTTAG
240 .|||||.|||||.||.||||||||||||||||||||.||.||.||.|||.|.||..|.||
BE037100. 295 GTCACTTTTAGTCCCTTACATATTCCTCAGTCTTCCATCCGGGCCATTTCATCTGTTCAG
gi|121904 577 GGGTGAGGTTGGGAAATGGATTGCTTTTGTCGCGGTTGTGCTAAGGCTATTCTTCCACCG
300 .||.|||||.||||||||||||||..|..|.||.||.|||.|||||||.||||||.||||
BE037100. 355 AGGCGAGGTCGGGAAATGGATTGCCATCATTGCAGTCGTGTTAAGGCTGTTCTTCAACCG
gi|121904 637 CCGCTTCCCAGAATGGTTAGAGATGCCAGGATCACTGATACTATTGTTGGTGGTAGCTCC
360 .|..|||||||..|||.|.||.|||||.|||||..|||||||..|..|||||||.||.||
BE037100. 415 GCATTTCCCAGTTTGGCTGGAAATGCCTGGATCGTTGATACTCCTCCTGGTGGTGGCACC
gi|121904 697 AGAATTGCTAGCACACAAATTAAAGGATAGTTGGATGGGAGTTGTAATTCTGTTAATCAT
420 |||.||..|..||||||||.|.|||||.||.|||||.|||.|||.||||.||.||...||
BE037100. 475 AGACTTCTTTACACACAAAGTGAAGGAGAGCTGGATCGGAATTGCAATTATGATAGCGAT
gi|121904 757 AGGGTGTTATTTGCTGCAAGAACATATCAGGGCAACTGGTGGTTTAAGAAATTCGTTTAC
480 |||||||.|..||.||||||||||||||||.||.||||||||.||..|.|||||.||.||
BE037100. 535 AGGGTGTCACCTGATGCAAGAACATATCAGAGCCACTGGTGGCTTTTGGAATTCCTTCAC
gi|121904 817 TCAAAGCCATGGAATTTCCTATACGATTGGGCTGCTTCTCTTATTGGCTTACCCAATTTG
540 .||.|||||.||||.||...|.||.||||||||..|.||..||.||||||||||..|.|-
BE037100. 595 ACAGAGCCACGGAACTTTTAACACAATTGGGCTTATCCTTCTACTGGCTTACCCTGTCT-
gi|121904 877 GTCCATGGTTATTTTCATGATTTA 901
600 ||..|||||.||.||||||||.|| 624
BE037100. 654 GTTTATGGTCATCTTCATGATGTA 678
count = 0
for hit in blast_record:
for alignment in hit:
print(alignment.target.id)
print(alignment.annotations['evalue'])
count += 1
if(count==5):
breakgi|1219041180|ref|XM_021875076.1|
6.36186e-117
gi|2514617377|ref|XM_021992092.2|
1.70712e-111
gi|2518612504|ref|XM_010682658.3|
4.58085e-106
gi|2031543140|ref|XM_041168865.1|
1.59887e-105
gi|2618480339|ref|XM_048479995.2|
5.58062e-105for hit in blast_record:
for alignment in hit:
if alignment.annotations['evalue'] < 1e-105:
print(alignment.target.description)
print(alignment[:,:50])PREDICTED: Chenopodium quinoa cold-regulated 413 plasma membrane protein 2-like (LOC110697660), mRNA
gi|121904 277 ACCGAAAATGGGCAGAGGAGTGAATTATATGGCAATGACACCTGAGCAAC 327
0 ||.|||||||||.||||.|.||||.||..||||.||||.|.||||.|||. 50
BE037100. 58 ACAGAAAATGGGGAGAGAAATGAAGTACTTGGCCATGAAAACTGATCAAT 108
PREDICTED: Spinacia oleracea cold-regulated 413 plasma membrane protein 2-like (LOC110787470), mRNA
gi|251461 41 AAAATGGGTAGACGAATGGATTATTTGGCGATGAAAACCGAGCAATTAGC 91
0 ||||||||.|||..||||.|.||.|||||.||||||||.||.|||||.|| 50
BE037100. 62 AAAATGGGGAGAGAAATGAAGTACTTGGCCATGAAAACTGATCAATTGGC 112
PREDICTED: Beta vulgaris subsp. vulgaris cold-regulated 413 plasma membrane protein 2 (LOC104895996), mRNA
gi|251861 24 TTGGCCATGAAAACTGAGCAAATGGCGTTGGCTAATTTGATAGATTATGA 74
0 |||||||||||||||||.|||.||||..||||||||.||||.||||..|| 50
BE037100. 86 TTGGCCATGAAAACTGATCAATTGGCCGTGGCTAATATGATCGATTCCGA 136
The hsps object has several attributes including the BLAST statistics such as evalue, score, positives, etc. These can be used to extract hits based on certain conditions. E.g., the code below shows saving hits from the previous Blast search with evalue greater than 1e-105 to a new file.
with open("new_file.txt", "w") as file_handle:
for hit in blast_record:
for alignment in hit:
if alignment.annotations['evalue'] < 1e-105:
print(alignment.target.id)
file_handle.write(str(alignment)+"\n")
file_handle.write("\n")
print("DONE")gi|1219041180|ref|XM_021875076.1|
gi|2514617377|ref|XM_021992092.2|
gi|2518612504|ref|XM_010682658.3|
DONEThe better approach to work with BLAST results is to create a dataframe having all the attributes for all the hits. The following code iterates through the BLAST output and creates a pandas dataframe having all the results.
import pandas as pd
blast_data = []
for hit in blast_record:
for alignment in hit:
blast_data.append({
'Score': alignment.score,
'Target ID': alignment.target.id,
'Target description': alignment.target.description,
'Bit score': alignment.annotations['bit score'],
'E-value': alignment.annotations['evalue'],
'identity': alignment.annotations['identity'],
'positives': alignment.annotations['positive'],
'gaps': alignment.annotations['gaps'],
})
df = pd.DataFrame(blast_data)
df.head()| Score | Target ID | Target description | Bit score | E-value | identity | positives | gaps | |
|---|---|---|---|---|---|---|---|---|
| 0 | 482.0 | gi|1219041180|ref|XM_021875076.1| | PREDICTED: Chenopodium quinoa cold-regulated 4... | 435.898 | 6.361860e-117 | 473 | 473 | 4 |
| 1 | 463.0 | gi|2514617377|ref|XM_021992092.2| | PREDICTED: Spinacia oleracea cold-regulated 41... | 418.766 | 1.707120e-111 | 447 | 447 | 3 |
| 2 | 443.0 | gi|2518612504|ref|XM_010682658.3| | PREDICTED: Beta vulgaris subsp. vulgaris cold-... | 400.732 | 4.580850e-106 | 448 | 448 | 4 |
| 3 | 441.0 | gi|2031543140|ref|XM_041168865.1| | PREDICTED: Juglans microcarpa x Juglans regia ... | 398.929 | 1.598870e-105 | 448 | 448 | 10 |
| 4 | 439.0 | gi|2618480339|ref|XM_048479995.2| | PREDICTED: Ziziphus jujuba cold-regulated 413 ... | 397.126 | 5.580620e-105 | 448 | 448 | 12 |
df.columnsIndex(['Score', 'Target ID', 'Target description', 'Bit score', 'E-value',
'identity', 'positives', 'gaps'],
dtype='object')Now that we have a dataframe, we can easily select hits based on desired thresholds. For instance, the code below selects all the hits with evalue less than 1e-105.
df[df["E-value"] < 1e-105]| Score | Target ID | Target description | Bit score | E-value | identity | positives | gaps | |
|---|---|---|---|---|---|---|---|---|
| 0 | 482.0 | gi|1219041180|ref|XM_021875076.1| | PREDICTED: Chenopodium quinoa cold-regulated 4... | 435.898 | 6.361860e-117 | 473 | 473 | 4 |
| 1 | 463.0 | gi|2514617377|ref|XM_021992092.2| | PREDICTED: Spinacia oleracea cold-regulated 41... | 418.766 | 1.707120e-111 | 447 | 447 | 3 |
| 2 | 443.0 | gi|2518612504|ref|XM_010682658.3| | PREDICTED: Beta vulgaris subsp. vulgaris cold-... | 400.732 | 4.580850e-106 | 448 | 448 | 4 |
To save the filtered set of hits (or the entire output) to a csv file, we can use the to_csv function.
df_top_hits = df[df["E-value"] < 1e-105]
df_top_hits.to_csv("Blast_hits.csv", index=False)# %load Blast_hits.csv
Score,Target ID,Target description,Bit score,E-value,identity,positives,gaps
482.0,gi|1219041180|ref|XM_021875076.1|,"PREDICTED: Chenopodium quinoa cold-regulated 413 plasma membrane protein 2-like (LOC110697660), mRNA",435.898,6.36186e-117,473,473,4
463.0,gi|2514617377|ref|XM_021992092.2|,"PREDICTED: Spinacia oleracea cold-regulated 413 plasma membrane protein 2-like (LOC110787470), mRNA",418.766,1.70712e-111,447,447,3
443.0,gi|2518612504|ref|XM_010682658.3|,"PREDICTED: Beta vulgaris subsp. vulgaris cold-regulated 413 plasma membrane protein 2 (LOC104895996), mRNA",400.732,4.58085e-106,448,448,4To run the Blast search using a sequence file instead of gi number, we first need to create a seqeunce object and then pass it on to the qblast function as shown below. To run this code, save the protein sequence below in a new file example1.fasta.
%load example1.fasta
seq_file = SeqIO.read('example1.fasta', 'fasta')
result_handle2 = Blast.qblast("blastp", "nr", seq_file.seq)
with open("test_blast.xml", "wb") as out_handle:
out_handle.write(result_handle2.read())
result_handle2.close()
blast_output = open("test_blast.xml", "rb")
blast_record = Blast.read(blast_output)
print(blast_record[0][0])Query : Query_4888877 Length: 570 Strand: Plus
unnamed protein product
Target: sp|P45897.1| Length: 570 Strand: Plus
RecName: Full=Dwarfin sma-4; AltName: Full=MAD protein homolog 3
[Caenorhabditis elegans] >gb|AAA97605.1| SMA-4 [Caenorhabditis elegans]
Score:1192 bits(3084), Expect:0,
Identities:570/570(100%), Positives:570/570(100%), Gaps:0.570(0%)
sp|P45897 0 MFHPGMTSQPSTSNQMYYDPLYGAEQIVQCNPMDYHQANILCGMQYFNNSHNRYPLLPQM
0 ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Query_488 0 MFHPGMTSQPSTSNQMYYDPLYGAEQIVQCNPMDYHQANILCGMQYFNNSHNRYPLLPQM
sp|P45897 60 PPQFTNDHPYDFPNVPTISTLDEASSFNGFLIPSQPSSYNNNNISCVFTPTPCTSSQASS
60 ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Query_488 60 PPQFTNDHPYDFPNVPTISTLDEASSFNGFLIPSQPSSYNNNNISCVFTPTPCTSSQASS
sp|P45897 120 QPPPTPTVNPTPIPPNAGAVLTTAMDSCQQISHVLQCYQQGGEDSDFVRKAIESLVKKLK
120 ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Query_488 120 QPPPTPTVNPTPIPPNAGAVLTTAMDSCQQISHVLQCYQQGGEDSDFVRKAIESLVKKLK
sp|P45897 180 DKRIELDALITAVTSNGKQPTGCVTIQRSLDGRLQVAGRKGVPHVVYARIWRWPKVSKNE
180 ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Query_488 180 DKRIELDALITAVTSNGKQPTGCVTIQRSLDGRLQVAGRKGVPHVVYARIWRWPKVSKNE
sp|P45897 240 LVKLVQCQTSSDHPDNICINPYHYERVVSNRITSADQSLHVENSPMKSEYLGDAGVIDSC
240 ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Query_488 240 LVKLVQCQTSSDHPDNICINPYHYERVVSNRITSADQSLHVENSPMKSEYLGDAGVIDSC
sp|P45897 300 SDWPNTPPDNNFNGGFAPDQPQLVTPIISDIPIDLNQIYVPTPPQLLDNWCSIIYYELDT
300 ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Query_488 300 SDWPNTPPDNNFNGGFAPDQPQLVTPIISDIPIDLNQIYVPTPPQLLDNWCSIIYYELDT
sp|P45897 360 PIGETFKVSARDHGKVIVDGGMDPHGENEGRLCLGALSNVHRTEASEKARIHIGRGVELT
360 ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Query_488 360 PIGETFKVSARDHGKVIVDGGMDPHGENEGRLCLGALSNVHRTEASEKARIHIGRGVELT
sp|P45897 420 AHADGNISITSNCKIFVRSGYLDYTHGSEYSSKAHRFTPNESSFTVFDIRWAYMQMLRRS
420 ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Query_488 420 AHADGNISITSNCKIFVRSGYLDYTHGSEYSSKAHRFTPNESSFTVFDIRWAYMQMLRRS
sp|P45897 480 RSSNEAVRAQAAAVAGYAPMSVMPAIMPDSGVDRMRRDFCTIAISFVKAWGDVYQRKTIK
480 ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Query_488 480 RSSNEAVRAQAAAVAGYAPMSVMPAIMPDSGVDRMRRDFCTIAISFVKAWGDVYQRKTIK
sp|P45897 540 ETPCWIEVTLHRPLQILDQLLKNSSQFGSS 570
540 |||||||||||||||||||||||||||||| 570
Query_488 540 ETPCWIEVTLHRPLQILDQLLKNSSQFGSS 570
Let’s say we need only the alignment with the mouse sequence, then, to print first 50 characters of each alignment with the mouse sequence along with corresponding statistics, the following code can be used.
for hits in blast_record:
for alignment in hits:
if "brenneri" in alignment.target.description:
print(alignment.target.description)
print(alignment.score, alignment.annotations['evalue'])unnamed protein product [Caenorhabditis brenneri]
2161.0 0.0
hypothetical protein CAEBREN_31444 [Caenorhabditis brenneri]
1889.0 0.0
sma-4, partial [Caenorhabditis brenneri]
997.0 1.39233e-129The GenomeDiagram Link module within Biopython’s Bio.Graphics package can be used to generate publication-quality representations of circular and linear genomes. The diagrams can be saved in different formats including PDF, PNG, and SVG. To use the GenomeDiagram module, the reportlab and pycairo libraries are required. These can be installed using the following command:
pip install "reportlab[pycairo]"
The code below shows creating a linear genome diagram for a plasmid sequence read from a GenBank file. The features of type “gene” are added to the diagram with different colors based on the strand information. The diagram is then saved in the png format. For this example, we’ll use a GenBank file from the Biopython tutorial repository available.
record = SeqIO.read("NC_005816.gb", "genbank")
recordSeqRecord(seq=Seq('TGTAACGAACGGTGCAATAGTGATCCACACCCAACGCCTGAAATCAGATCCAGG...CTG'), id='NC_005816.1', name='NC_005816', description='Yersinia pestis biovar Microtus str. 91001 plasmid pPCP1, complete sequence', dbxrefs=['Project:58037'])for feature in record.features:
if feature.type == "gene":
print(feature.location)[86:1109](+)
[1105:1888](+)
[2924:3119](+)
[3485:3857](+)
[4342:4780](+)
[4814:5888](-)
[6004:6421](+)
[6663:7602](+)
[7788:8088](-)
[8087:8360](-)To create a genome diagram, we’ll first initialize a Diagram object followed by a Track and a FeatureSet to hold the features to be added to the diagram.
from Bio.Graphics import GenomeDiagram
from reportlab.lib import colors
from reportlab.lib.units import cm
gd_diagram = GenomeDiagram.Diagram(record.description)
gd_track_for_features = gd_diagram.new_track(1, name="Annotated Features")
gd_feature_set = gd_track_for_features.new_set()Next, we’ll add features to the gd_feature_set object using the add_feature method. The features will be colored differently based on their position and strand information.
for feature in record.features:
if feature.type == "gene":
if len(gd_feature_set) % 2 == 0:
color = colors.blue
else:
color = colors.pink
if feature.location.strand == -1:
color = colors.green
gd_feature_set.add_feature(feature, color=color, label=True)Finally, we’ll draw the diagram and save it in the png format. The franments argument in the draw function specifies the number of fragments of the genome to create when rendering the diagram.
gd_diagram.draw(
format="linear",
orientation="landscape",
pagesize="A4",
fragments=4,
start=0,
end=len(record),
)
gd_diagram.write("plasmid_linear.png", "PNG", dpi=300)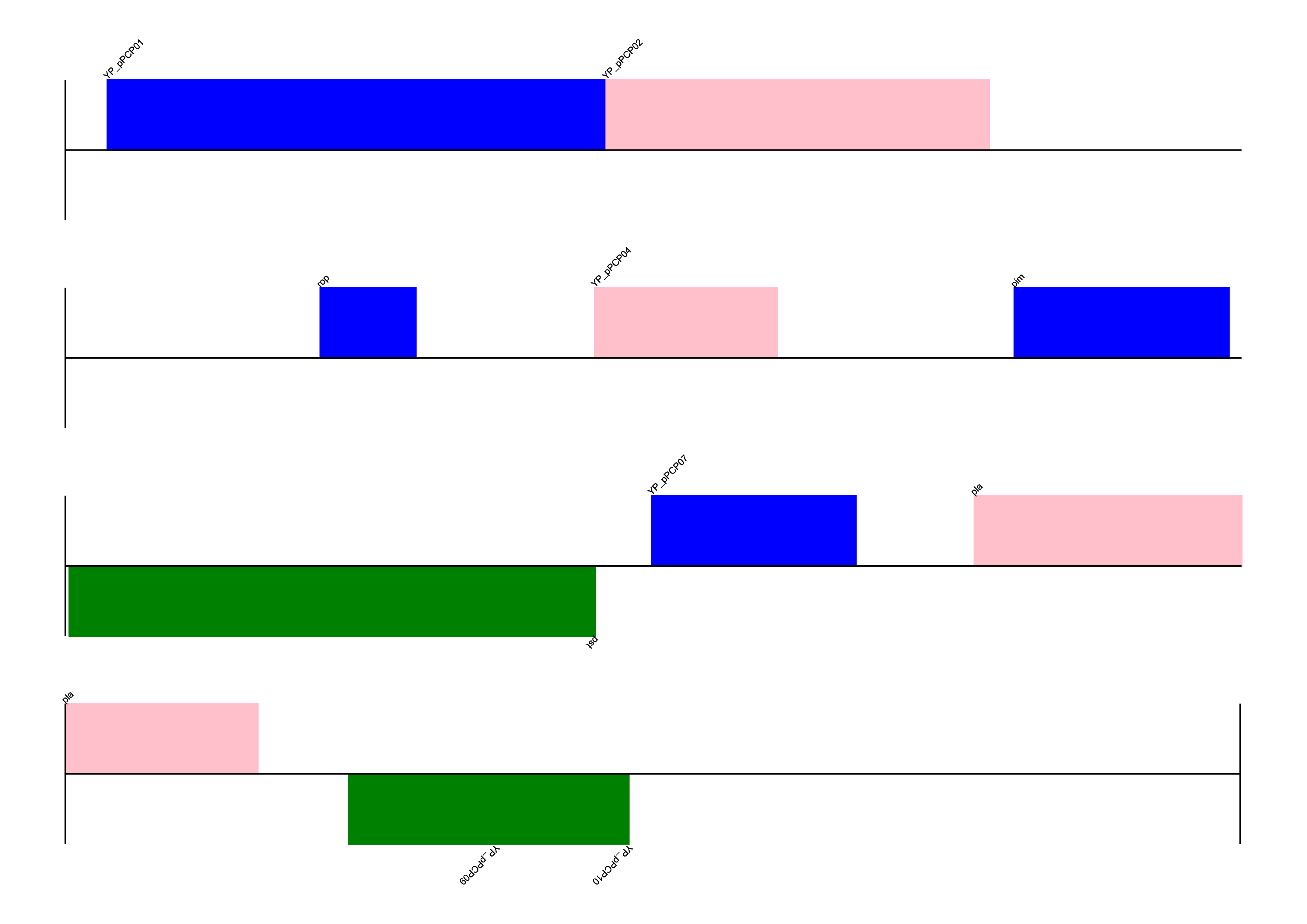
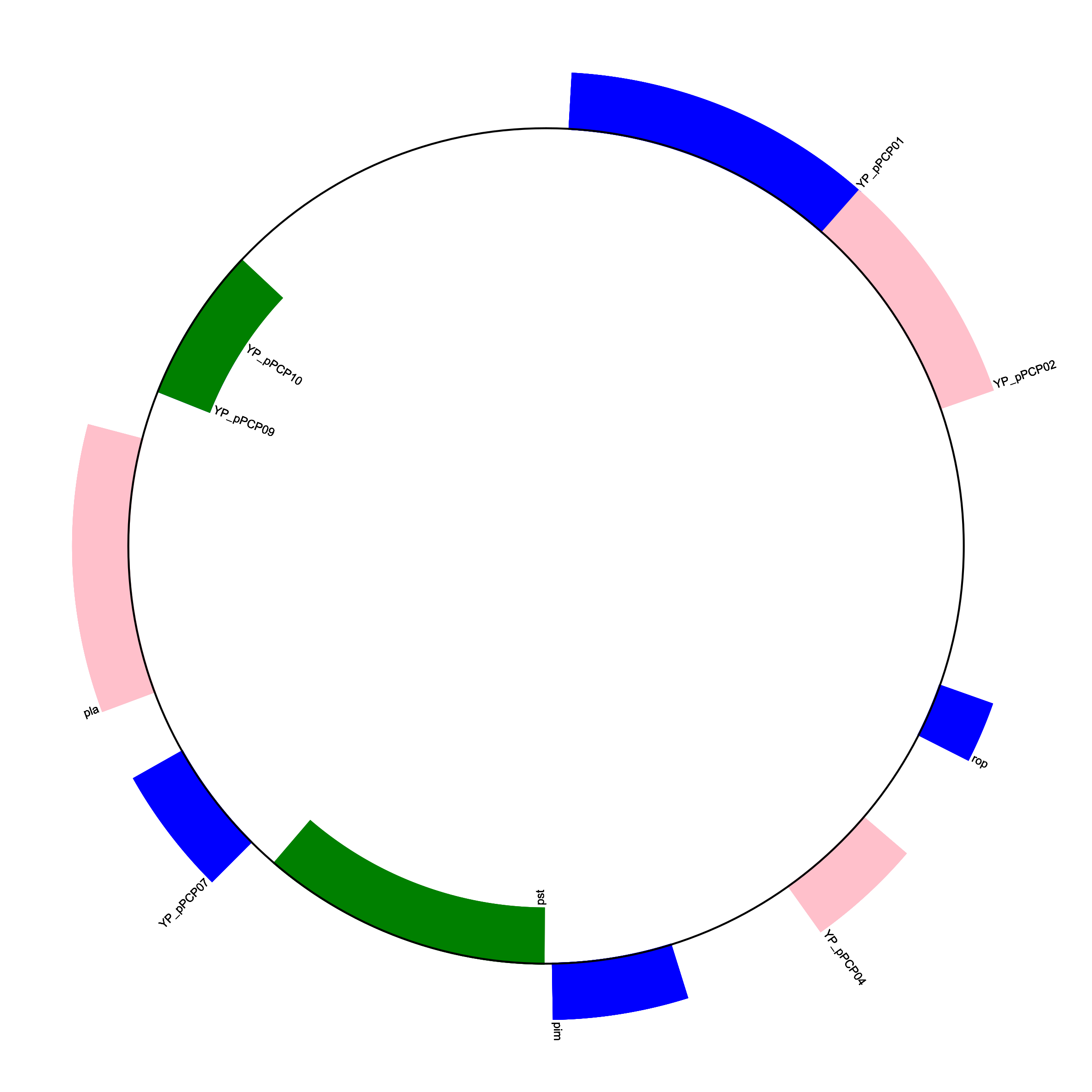
Similarly, we can create a circular genome diagram by specifying the format keyword argument as circular within the draw function.
gd_diagram.draw(
format="circular",
circular=True,
pagesize=(20 * cm, 20 * cm),
start=0,
end=len(record),
circle_core=0.7,
)
gd_diagram.write("plasmid_circular.png", "PNG", dpi=300)